
See what API testing solution came out on top in the GigaOm Radar Report. Get your free analyst report >>
Jump to Section
Navigating FDA Validation Guidance for Medical Device Cybersecurity

November 13, 2023
13 min read
With the FDA’s addition of more requirements to their cybersecurity validation and standards comes the need for medical software manufacturers to adopt static analysis to ensure that their software meets these new security standards. Read on to learn how to implement static analysis to meet these security requirements.
Jump to Section
Jump to Section
As the FDA adds more cybersecurity requirements to their software validation guidance, medical device manufacturers can turn to static analysis, the most effective method to address safety and security concerns and deliver predictable software.
Medical device manufacturers continue to focus on improving software development processes for two primary reasons.
- Address growing security threats.
- Satisfy the FDA’s requirements, which are becoming more precise.
Understanding the New FDA Cybersecurity Guidance
The FDA’s new cybersecurity guidance for medical devices was released in September 2023 and provides comprehensive recommendations to the industry regarding cybersecurity device design, labeling, and documentation to be included in premarket submissions for devices with cybersecurity risk.
The guidance is intended to help manufacturers identify and mitigate cybersecurity risks throughout the life cycle of a medical device, from design and development to manufacturing, servicing, and post market surveillance. It also provides recommendations for communicating cybersecurity risks to healthcare providers and patients.
The new guidance builds on the FDA’s previous cybersecurity guidance documents, but it also includes a number of new and updated recommendations, such as:
- A requirement for manufacturers to implement a comprehensive cybersecurity risk management program.
- A requirement for manufacturers to submit a software bill of materials (SBOM) for their devices.
- A requirement for manufacturers to develop and implement cybersecurity plans for post market support.
- Recommendations for communicating cybersecurity risks to healthcare providers and patients.
The FDA used to be focused on functional safety aspects of the systems, but now cybersecurity is a subject of equal importance. Even though safety and security are similar—and you could easily argue that both are about creating predictable software—the FDA considers cybersecurity something that requires dedicated attention and measures.
The Importance of Process Validation in Medical Devices
Process validation is the collection and evaluation of data that establishes scientific evidence that a process is capable of consistently delivering a quality product. In the context of medical devices, process validation is essential for ensuring the safety and effectiveness of devices throughout their life cycle.
Process validation is particularly important for medical devices that are controlled by software, as software can be complex and difficult to test comprehensively. For example, a software-controlled medical device may have millions of lines of code, and it’s not possible to test every possible scenario and combination of inputs and outputs without utilizing both static code analysis and dynamic analysis.
Below are three key reasons why process validation matters.
- Quality assurance. Process validation helps ensure that medical devices are manufactured consistently and reliably, reducing the risk of defects and ensuring a high level of quality.
- Patient safety. By validating the manufacturing process, potential risks and defects can be identified and mitigated, enhancing patient safety.
- Compliance with regulatory requirements. Meeting regulatory requirements, such as the FDA’s Quality System Regulation (QSR), necessitates the use of validated processes. Failing to do so can lead to compliance issues.
Regulatory Requirements and the FDA Guidance Document
The regulatory requirements outlined in the FDA guidance document on medical device cybersecurity, are designed to address the growing concerns regarding the security and safety of medical devices in an increasingly connected and digital healthcare environment. Here’s an explanation of the regulatory requirements.
Statutory Authority
The FDA now has new statutory authority under the Food and Drug Omnibus Reform Act (FDORA), signed into law in December 2022. This law allows the FDA to require cybersecurity information in medical device submissions for “cyber devices” and mandates that manufacturers take specific actions to demonstrate reasonable assurance that their devices and related systems are “cybersecure.” Failure to comply with these requirements is considered a prohibited act, subject to prosecution.
Definition of “Cyber Device”
The term “cyber device” is newly defined in the law as a device that includes software validated, installed, or authorized by the sponsor, can connect to the internet, and contains technological characteristics that could be vulnerable to cybersecurity threats. This definition helps clarify which devices are subject to the new cybersecurity requirements.
Inclusion of Cybersecurity Information in Premarket Submissions
The new guidance mandates that manufacturers must include specific cybersecurity information in their premarket submissions to the FDA. These premarket submissions can include Premarket Approval (PMA), 510(k), or de novo submissions. The cybersecurity information is required to ensure that the device meets the cybersecurity requirements outlined in the law.
Monitoring and Addressing Post Market Cybersecurity Vulnerabilities
Device manufacturers are required to submit a plan for monitoring, identifying, and addressing post market cybersecurity vulnerabilities and exploits. This plan should outline the manufacturer’s approach to continuously monitoring and addressing cybersecurity risks that may emerge after the device is on the market.
Reasonable Assurance of Cybersecurity
Manufacturers must design, develop, and maintain processes and procedures to provide reasonable assurance that the device and its related systems are “cybersecure.” This requirement emphasizes the need for a proactive approach to building security into the device’s design and maintaining it throughout the device’s life cycle.
Software Bill of Materials
Manufacturers are expected to provide a software bill of materials (SBOM) as part of their submissions. This bill should detail the commercial, open-source, and off-the-shelf software components used in the device. This information is crucial for identifying and addressing potential vulnerabilities in the software stack.
Compliance With Additional FDA Requirements
The guidance document also empowers the FDA to establish additional requirements through regulations to demonstrate that the device and related systems are cybersecure. Manufacturers must be prepared to comply with any such additional requirements as outlined by the FDA.
Prohibited Act for Noncompliance
A key aspect of the regulatory requirements is the inclusion of a new statutory prohibited act. This means that failure to comply with the FDA’s cybersecurity requirements is not just a compliance issue but also a legal violation. The government has the authority to prosecute violations of these requirements criminally or pursue injunctive relief against noncompliant companies.
How Medical Device Manufacturers Can Comply
Medical device manufacturers must comply with many requirements to ensure the safety, efficacy, and quality of their products. We’ll explore key aspects of compliance with FDA regulatory requirements in medical devices, laying more emphasis on addressing security concerns in medical devices and designing controls and validation protocols for manufacturers.
Addressing Security Concerns in Medical Devices
To comply with FDA regulations and prioritize patient safety, medical device manufacturers must address security concerns effectively. Here are some tips to address security concerns in medical devices:
- Standardize your code and conduct a risk assessment. The first step to having a medical software device devoid of security issues is to have the software written with standardized coding rules. For already existing software, you can check and clean up the code base to ensure it complies with acceptable coding standards. Medical device manufacturers can leverage static code analysis solutions to test and analyze the source code of their software programs. In addition to static code analysis, other types of software testing that can help to deliver great software include integration testing, security testing, unit testing, system testing and usability testing. Cleaning the software code base and conducting risk analysis helps manufacturers identify potential security vulnerabilities in their devices. This includes assessing risks related to data breaches, unauthorized access, and the integrity of device functionality.
- Software updates and patch management. Regularly update and patch device software to address known vulnerabilities. Timely updates help in preventing security breaches that may exploit outdated software.
- Secure data storage. Ensure that patient data is stored securely, both on the device and during data transmission. Utilize secure data storage practices and industry-recommended encryption protocols to protect patient privacy and comply with data protection regulations.
- Collaborate with cybersecurity experts. Consider collaborating with cybersecurity experts and professionals who specialize in medical device security. Their expertise can help manufacturers identify and mitigate potential security risks.
Design Controls and Validation Protocol for Manufacturers
Proper design controls and validation protocols are vital components of ensuring that medical devices meet FDA regulatory requirements. These controls help manufacturers develop safe and effective devices that deliver the intended benefits to patients.
Here are key considerations for implementing design controls and validation protocols.
- Design and development planning. Manufacturers should establish a clear design and development plan that outlines the scope, objectives, and expectations for the medical device. This plan should include risk management, usability studies, and regulatory compliance.
- Design inputs and outputs. Define design inputs that specify the device’s requirements and design outputs that describe the device’s characteristics. Ensure that the design inputs align with user needs and regulatory requirements.
- Verification and validation. Implement rigorous verification and validation processes to confirm that the device meets its design requirements and intended use. This includes testing and evaluating the device’s performance and safety.
- Design history file (DHF). Maintain a comprehensive DHF that records all design and development activities, including documentation of design controls, test results, and design changes.
- Regulatory submissions. Submitting accurate and complete premarket notifications (510(k)), De Novo requests or premarket approval applications (PMA) is essential for gaining FDA clearance or approval for your medical devices. These submissions provide the FDA with necessary information about your device’s safety and efficacy, helping them assess its suitability for the market. Failure to complete these submissions correctly can lead to delays or even rejection of your product.
- Postmarket surveillance (PMS). Establishing a PMS system is critical for monitoring and reporting adverse events, complaints, and device malfunctions. Compliance with PMS requirements helps identify safety issues and implement corrective actions swiftly. It ensures that manufacturers are actively monitoring their devices’ performance and addressing any problems arising from use.
Tools and Methods for FDA Compliance
Manufacturers can employ various tools and methods to achieve and maintain compliance with FDA regulations for medical devices. Discussed below are essential tools and methods that can aid in the pursuit of FDA compliance.
Static Analysis for Meeting FDA Requirements
Static analysis plays a critical role in helping medical device manufacturers meet the stringent requirements set forth by the FDA.
Here are 5 reasons to adopt static analysis for meeting FDA requirements.
- Early issue detection. Static analysis tools can identify potential issues such as coding errors, security vulnerabilities, and design flaws at an early stage in the development process. This early detection allows for timely corrections, reducing the risk of costly and time-consuming modifications later in the development cycle. Recommended coding standards for C and C++ are MISRA C/C++ and CERT C/C++.
- Improved software quality. High-quality software is imperative for the safety and effectiveness of medical devices. Static analysis aids developers in creating cleaner, more reliable, and more secure code. This not only facilitates FDA compliance but also mitigates the risk of post market issues and safety concerns.
- Cybersecurity compliance. Given the increasing importance of cybersecurity for medical devices, the FDA places a strong emphasis on it. Static analysis tools can identify security weaknesses and vulnerabilities in the software, ensuring alignment with FDA guidance on cybersecurity.
- Regulatory documentation. Static analysis can provide comprehensive documentation and reports that demonstrate the thorough examination of the software and the steps taken to address any identified issues. This documentation is essential for FDA submissions and inspections.
- Streamlined validation. By identifying and addressing potential issues early, static analysis can expedite the validation process, which is an important step in FDA compliance. This helps reduce delays in bringing medical devices to market.
How to Ensure Proper Risk Management
Given the place of risk management in achieving the overall objectives or requirements of the FDA, let’s delve into how you can achieve proper risk management.
- Risk assessment. Risk assessment in medical device manufacturing is the initial step in identifying and understanding potential hazards associated with a medical device. Its purpose is to systematically evaluate the device’s design, intended use, biocompatibility, and other risk factors to pinpoint potential risks. A risk assessment exercise typically involves forming a cross-functional team with diverse expertise, including engineers, clinicians, and regulatory specialists.
- Risk analysis. Risk analysis aims to evaluate and prioritize the identified risks by assigning a risk score based on the probability of occurrence and the severity of potential harm. Risk analysis involves quantifying each risk by assessing its likelihood and potential impact. Depending on your company, a risk matrix or other scoring system can be used to prioritize risks. High-risk items are given priority for further action.
- Risk mitigation. Risk mitigation involves the development of strategies to reduce or control identified risks to an acceptable level. It aims to enhance the safety and efficacy of the medical device. Mitigation strategies can take various forms, including design modifications, safety features, better materials, enhanced user training, or improved instructions for use. The goal is to address the identified risks in a way that minimizes their impact.
- Documentation. Proper documentation is essential for transparency, accountability, and regulatory compliance. It provides a record of the risk management process and helps demonstrate due diligence to regulatory authorities. All aspects of the risk management process, including risk assessments, analyses, and mitigation plans, should be meticulously documented. This documentation is maintained throughout the product’s life cycle.
- Continuous improvement. The risk management process should be a dynamic and evolving system. Continuous improvement ensures that your risk management practices remain effective in addressing new risks and changing market conditions. Regular reviews and updates are conducted to incorporate lessons learned from past projects and adapt to evolving technology and regulations. This may involve revisiting and revising risk assessments, analyses, and mitigation plans.
How Static Analysis for C/C++ Can Help With Risk Management
Many of our medical device customers, when starting from the ground up, have found success introducing static analysis for C/C++ by following these steps:
- Review existing guidelines in the organization. Even if designed to be enforced manually, map them as much as possible to the checkers that are provided with the static analysis tool. A mature static analysis tool will likely cover most of them. You can consider building custom checkers for the remaining guidelines that couldn’t be mapped to static analysis checkers right away.
- Review the popular coding standards, especially those created with security in mind. Select a subset of guidelines for your team to follow. When selecting the guidelines, it makes sense to follow the categorization from the standards and select the guidelines that are categorized as the most important. For example, in CERT you probably want to start with L1 guidelines, and in MISRA C 2023, you will want to review mandatory guidelines.
- Define the static analysis tool configuration, and include your organization-specific guidelines and selected guidelines such as CERT. Do not enable all checkers at once. Instead, start with a small subset to avoid flooding developers with violations.
- Make sure your developers can scan their code immediately after it is created. It also makes sense to include static analysis in the CI/CD process.
- As the developers make progress and clean the source code, make sure to progressively enable more checkers from your list. At the end of the day, you want to provide evidence that you comply with the entire set of guidelines you selected not that you stopped halfway through. To better orient yourself in the process, and understand your current progress, you can deploy a central reporting system to help aggregate the testing data and monitor developers’ work. See an example of this below:
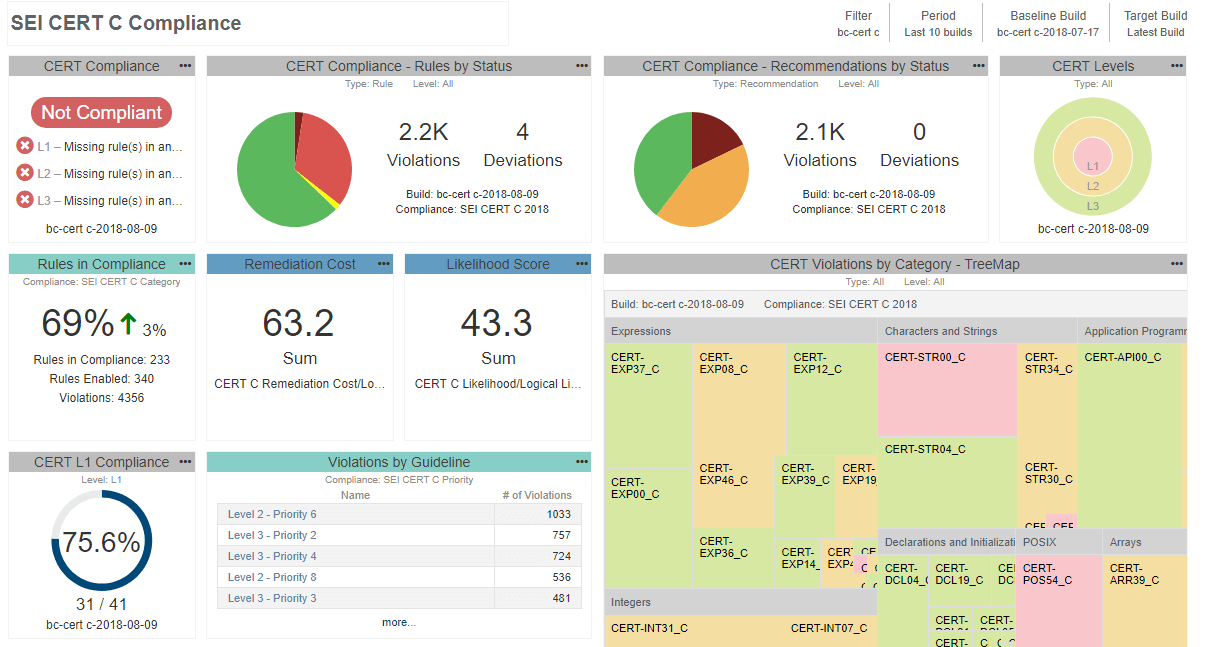
Since static analysis reports become part of the quality management system, you can’t use just any tool. FDA requires that all tools used in development and verification of the software be validated for intended use. There are different ways to demonstrate the tool’s suitability for use in safety-critical development. Depending on the risk of the device, it could be as simple as re-using a certificate of compliance or completing the lengthier process of tool qualification.
Using a TÜV SÜD Certification
For the end user, the most convenient option is to take the credit for the work done by the tool vendor and re-use the certification that is granted for the testing tool by an external certification organization such as TÜV SÜD. Parasoft C/C++test, for example, is covered with a TÜV SÜD certification that can be re-used to demonstrate suitability for developing software according to medical standards like IEC 62304.
Performing Tool Qualification
For high-risk devices such as Class C, you may need to validate the tool internally in your development environment. The intention is to provide the evidence that the tool operates according to its operational requirements, gathered in the project’s development environment. This is a very tedious and time-consuming process.
The best situation is if your tool vendor can support you in this effort and provide a special tool qualification kit containing well-designed test cases, and the automation framework to execute them in the project’s development environment, and automatically generate the documentation that can serve as the evidence for tool validation. Here again, Parasoft’s flagship product C/C++test, provides an automated tool qualification kit.
Case Studies and Real-World World Applications
We have a plethora of case studies of real-world applications of Parasoft static analysis tools. But let’s look at two distinct success stories that exemplify how our static analysis solution has played a significant role in helping medical device manufacturers overcome their unique challenges and meet FDA requirements.
Medical Device Manufacturer Success Stories
The following case studies illustrate how Parasoft’s software testing solutions have been instrumental in helping medical device manufacturers address their unique challenges and achieve their goals. Inovytec and Smiths Medical both saw remarkable improvements in code quality, compliance, and testing efficiency through their collaboration with Parasoft.
Inovytec’s Journey to FDA 510(k) Certification
Inovytec, a company dedicated to producing medical devices, embarked on a mission to achieve FDA 510(k) certification for its Ventway Sparrow ventilator. Their challenge lay in delivering clean code while adhering to FDA regulations. Parasoft’s C/C++ static code analysis solution came to their rescue.
Inovytec’s software development team customized Parasoft C/C++test to align with the stringent FDA requirements. Every time they prepared to release a new software version, they ensured that Parasoft’s static analysis was configured to run according to the FDA regulation definitions. The result was not only improved code quality but also a resounding success in passing 100% of the FDA 510(k) certification rules and guidelines. Parasoft emerged as the preferred testing solution at Inovytec, and their collaboration with ESL, a distributor of Parasoft products in Israel, provided essential support and expertise whenever needed.
Smiths Medical’s Adoption of Test-Driven Development (TDD)
Smiths Medical, a renowned manufacturer of specialty medical devices, encountered a series of challenges in its quest to develop high-quality, safety-critical medical device software. Automated testing plays a crucial role in Smiths Medical’s testing strategy.
Past endeavors to incorporate tools fell short of complete success. The development team sought a solution that could enhance their entire testing process by embracing a fresh perspective centered around unit testing and test-driven development (TDD), a methodology that integrates design, testing, and code development. They needed a tool that could fit within their testing pipeline and enhance their overall development culture. Parasoft C/C++test proved to be the answer to their challenges.
The software team not only achieved successful TDD adoption but also benefited from improved test stability, enhanced code coverage, and a streamlined tool qualification process, which was critical for safety-critical applications. With Parasoft, Smiths Medical was able to transform its development processes, making testing an integral part of its software pipeline, and ultimately, ensuring the delivery of safe, high-quality medical devices.
Overcoming Challenges in FDA Validation
The FDA validation process can be complex and challenging for medical device manufacturers. However, there are a number of steps that manufacturers can take to overcome these challenges.
- Start early. The FDA validation process should start early in the product development cycle. This will give manufacturers more time to plan and execute the validation process, and it will help to identify and address any potential problems early on.
- Use a risk-based approach. The FDA validation process should be based on a risk assessment of the device. Manufacturers should focus their validation efforts on the most critical components and functions of the device.
- Use qualified personnel. The FDA validation process should be conducted by qualified personnel who have experience with FDA regulations and validation best practices.
- Document the validation process. Manufacturers should document the FDA validation process thoroughly. This will help manufacturers to demonstrate to the FDA that they have validated their device in accordance with FDA regulations.
Next Steps for Navigating FDA Validation Guidance
The FDA provides essential validation guidance for medical device manufacturers to ensure product quality and patient safety. As regulations and guidelines evolve, staying on top of these changes and planning for future compliance is crucial. Two ways medical device manufacturers can do this is to prepare for future FDA guidelines and remain compliant.
Preparing for Future FDA Guidelines
The FDA is constantly updating and revising its guidance documents for medical device manufacturers. To prepare for future FDA guidelines, medical device manufacturers should:
- Monitor the FDA’s website for updates to guidance documents. The FDA publishes updates to its guidance documents on its website. Manufacturers should regularly check the FDA’s website for updates to guidance documents that are relevant to their devices.
- Participate in FDA workshops and webinars. The FDA frequently hosts workshops and webinars on a variety of topics related to medical device regulation. Manufacturers should participate in these events to stay up-to-date on the latest FDA thinking on medical device regulation.
- Network with other medical device manufacturers. Manufacturers can network with other medical device manufacturers to share information and best practices for complying with FDA regulations.
Ensuring Continuous Compliance
Medical device manufacturers must ensure continuous compliance with FDA regulations. To do this, manufacturers should:
- Establish and maintain a quality system. A quality system is a set of procedures and processes that manufacturers use to ensure the safety and effectiveness of their devices. Manufacturers should establish and maintain a quality system that meets the requirements of the FDA’s Quality System Regulation.
- Conduct regular audits of their quality system. Manufacturers should conduct regular audits of their quality system to identify and address any areas of noncompliance.
- Submit MDR reports to the FDA. Manufacturers are required to submit Medical Device Reports (MDR) to the FDA for adverse events that occur with their devices. By submitting MDR reports to the FDA, manufacturers can help the FDA identify and address safety concerns with their devices.
Over time, achieving FDA compliance has proven to be quite rigorous and time-consuming. However, with static analysis and dynamic analysis tools, these challenges can be overcome. Static analysis tools offer advanced static code analysis capabilities, which help identify and rectify issues in software and code early in the development process.
Introducing static analysis is a dedicated effort, requiring developer time and cost. But it’s a proven way to harden your system against malicious attacks. Deploying static analysis with a well-thought-out set of security guidelines enables you to build systems that can stand up against unforeseen future attacks.
IEC 62304 Software Compliance in the Medical Industry
“MISRA”, “MISRA C” and the triangle logo are registered trademarks of The MISRA Consortium Limited. ©The MISRA Consortium Limited, 2021. All rights reserved.
Recommended Content

